

Abstract
The SETD2 gene encodes the only methyltransferase responsible for histone H3 lysine 36 trimethylation (H3K36Me3) in humans. H3K36me3 play a key role in preserving the fidelity of transcription elongation and splicing. In addition, SETD2/H3K36me3 have more recently been implicated in the maintenance of genomic integrity by regulating homologous recombination (HR) repair, Mismatch Repair (MMR) mitotic spindle assembly and chromosome segregation. SETD2 deletions and/or inactivating mutations occur in many solid tumors and have recently been found also in acute leukemias. We have reported that the HMC-1.1 and -1.2 mast cell leukemia (MCL) cell lines and many advanced systemic mastocytosis (SM) patients (pts) display H3K36Me3 deficiency as a result of non-genomic loss of function of SETD2. Proteasome inhibition restored SETD2 protein expression and H3K36me3, suggesting that a functional protein is produced but rapidly degraded. In an attempt to uncover the mechanisms underlying this phenomenon, we used an in silico approach to identify candidate SETD2-interacting proteins, followed by experimental confirmation by co-immunoprecipitation (co-IP). We found that, after proteasomal inhibition, SETD2 co-immunoprecipitates with the ubiquitin E3 ligase MDM2. Treatment with the MDM2 inhibitor SP-141 rescued SETD2 expression and H3K36Me3, suggesting that MDM2 may play a role in SETD2 degradation in ASM and MCL. Moreover, SP-141 treatment of HMC-1 cells at micromolar doses induced cytostatic but not cytotoxic effects as shown by cell growth curves. Clonogenic assays supported the cytostatic effects of SP141 in HMC-1.1 and -1.2 cells. siRNA-mediated knock-down of MDM2 also rescued SETD2 expression and activity, further supporting the hypothesis that SETD2 hyper-ubiquitination by MDM2 plays a role in SETD2 reduced stability and proteasomal degradation. Co-IP also showed that SETD2 interacts with Aurora Kinase A, as it was suggested in silico. We found that Aurora A is overexpressed in advanced SM and may target SETD2 for phosphorylation. Both pharmacological inhibition by Danusertib and siRNA-mediated silencing of Aurora A rescued SETD2 expression and activity, raising the hypothesis that phosphorylation by Aurora A might be the trigger for MDM-2 mediated degradation of SETD2.
To evaluate whether increased DNA damage and reduced HR proficiency can be observed in SETD2/H3K36Me3-deficient SM, we used western blotting (WB) and immunofluorescence (IF) to assess phosphorylated histone 2A.X (γH2AX) and Rad51 foci. Compared to cells from healthy controls, SETD2- and H3K36Me3-deficient cell lines and pts had significantly higher levels of γH2AX and lower levels of Rad51. RNA-seq in SETD2-deficient pts showed evidence of transcription and splicing defects like transcription-induced chimeras, intron retention and non-canonical splicing patterns not observed in healthy donors.
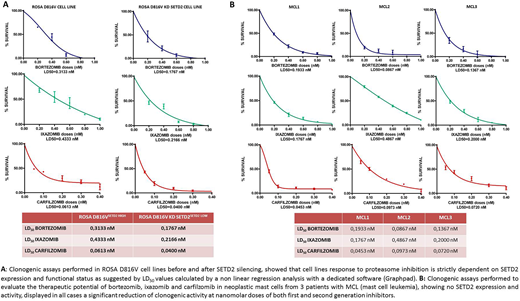
Next, the ROSAD816V cell line, which displays SETD2 and H3K36me3 levels superimposable to healthy donors, was studied by WB and IF to assess γH2AX and Rad51 in steady state and after sub-lethal DNA damage by UV exposure. The same experiments were carried out after SETD2 silencing for 2 months. Cells with silenced SETD2 had significantly higher levels of γH2AX and were unable to activate the HR repair. Interestingly, clonogenic assays in ROSAD816V cells before and after SETD2 silencing showed that reduction of clonogenic potential after proteasomal or MDM2 inhibition is indeed SETD2-dependent (Figure 1A).
Finally, we performed clonogenic assays to evaluate the therapeutic potential of bortezomib, carfilzomib and ixazomib in neoplastic mast cells from 3 patients with advanced SM and we observed in all cases that both first and second generation inhibitors induced a significant reduction of clonogenic activity at nanomolar doses (Figure 1B).
Taken together, our results suggest that AKA and MDM2-mediated post-translational modifications contribute to SETD2 non-genomic loss of function in advanced SM. Loss of SETD2 and H3K36me3 is associated with increased DNA damage and transcription and splicing defects in patients. Inhibiting AKA or MDM2 activity or proteasome-mediated degradation are promising therapeutic strategies in patients with low SETD2 expression levels.
Acknowledgments: study supported by AIRC (project code 16996) and AIL (Associazione Italiana contro le Leucemia, Linfomi e Mieloma).
Bonifacio:Incyte: Consultancy; Pfizer: Consultancy; Amgen: Consultancy; Novartis: Research Funding; Bristol Myers Squibb: Consultancy. Pagano:Janssen: Speakers Bureau; Merck: Speakers Bureau; Gilead: Speakers Bureau; Basilea: Speakers Bureau; Pfizer: Speakers Bureau. Valent:Novartis: Honoraria; Pfizer: Honoraria; Incyte: Honoraria. Cavo:Celgene: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau; Bristol-Myers Squibb: Honoraria, Membership on an entity's Board of Directors or advisory committees; AbbVie: Honoraria, Membership on an entity's Board of Directors or advisory committees; GlaxoSmithKline: Honoraria, Membership on an entity's Board of Directors or advisory committees; Adaptive Biotechnologies: Honoraria, Membership on an entity's Board of Directors or advisory committees; Takeda: Honoraria, Membership on an entity's Board of Directors or advisory committees; Janssen: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau; Amgen: Honoraria, Membership on an entity's Board of Directors or advisory committees. Soverini:Novartis: Consultancy; Incyte Biosciences: Consultancy; Bristol Myers Squibb: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal